
Research at NIAID’s Rocky Mountain Laboratories
Scientists at NIAID’s Rocky Mountain Laboratories (RML) in Hamilton, Montana, have studied prion diseases since the 1960s when Dr. William Hadlow spearheaded work on the sheep brain disease known as scrapie, which was later shown to be a prion disease. RML is one of the world's premier laboratories for studying prion diseases. Primary to their mission is understanding how abnormal prion protein cause disease at the molecular, biochemical, cellular, and animal-model levels.
Nervous System Interaction With Prion Protein
NIAID scientists at RML are studying how cells in the nervous system interact with prion protein and whether those interactions affect disease progression. These studies have shown how prions move through the brain and how cells in the brain called microglia help slow down disease.
Creutzfeldt-Jakob Disease (CJD) Studies
Other studies at RML have looked at different types of Creutzfeldt-Jakob disease (CJD) in human tissue and how different types of CJD may occur. These studies have shown how human prions interact with cells. They have also shown that prions derived from slightly different forms of the prion protein gene can influence how prions accumulate in the brain.
Prion Protein and Brain Injury
NIAID scientists at RML also have shown that, in response to damage to the brain, normal prion protein acquires properties similar to those of infectious prion protein. These studies show that prion protein, like the tau protein in chronic traumatic encephalopathy (CTE), acts abnormally following brain injury. They also suggest that damage to the brain might be a way in which CJD infection starts in some people.
Chronic Wasting Disease Studies
RML chronic wasting disease studies have focused on whether infectious prions can cross species from cervids, like deer and elk, into people. During research that took 13 years of observation, NIAID scientists published a series of studies, the latest in 2018, showing that CWD from deer or elk does not cause disease in prion models that are susceptible to infection by human prions. These studies reinforce the belief that a strong barrier to CWD infection exists between cervids and people.
Cerebral Organoid (“minibrain”) Cultures
Studies of prion disease infection of cerebral organoid (“minibrain”) cultures in incubators began at RML in 2017. These studies could provide a new model for scientists to study how prion diseases affect the human brain. Cerebral organoids are small balls of human brain cells – developed from skin stem cells – that range in size from a poppy seed to a small pea. Their organization, structure, and electrical signaling mimic some aspects of brain tissue.
Scientific Advances
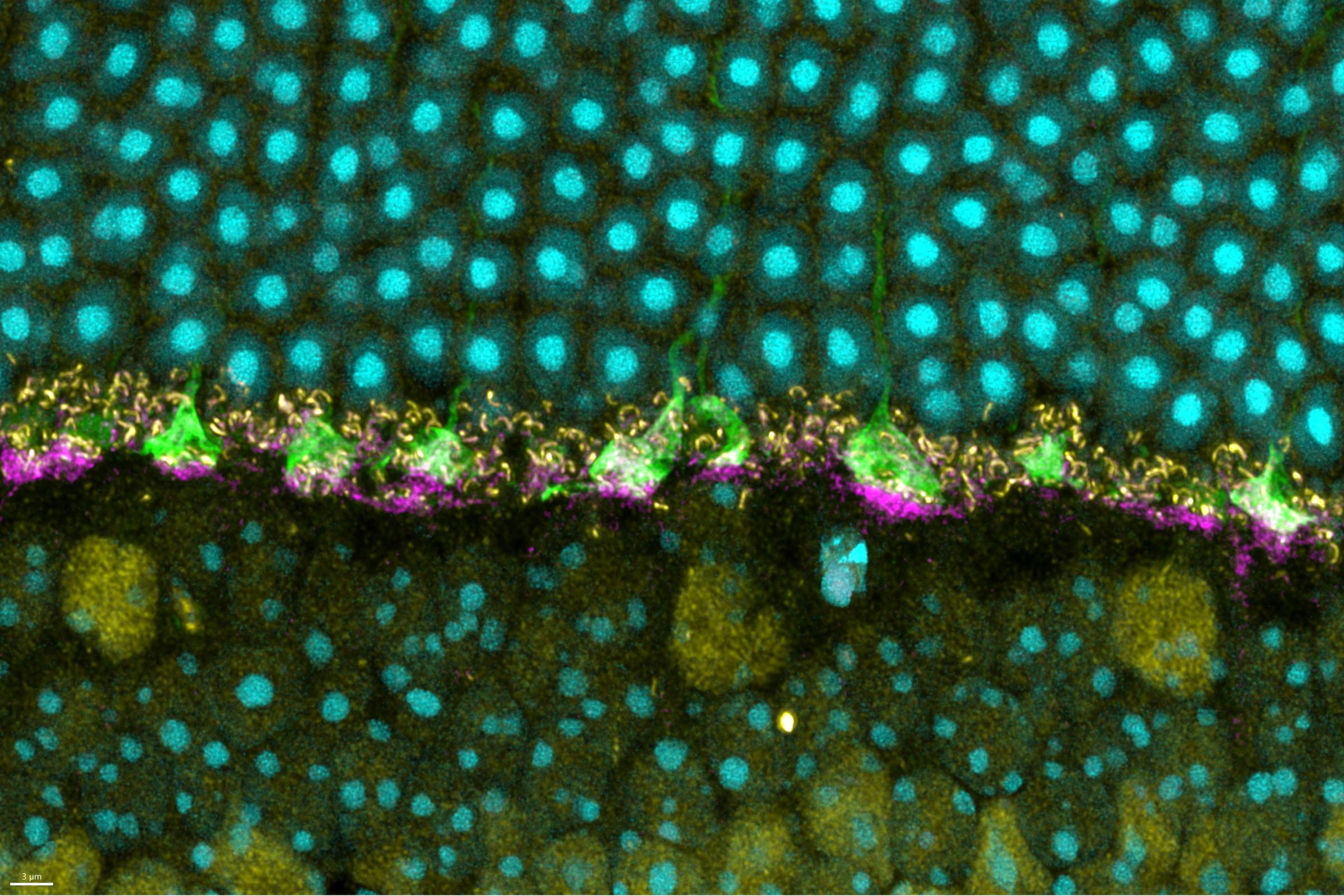
The Eyes Have it: A Functional Role for Prion Protein
September 26, 2024
Answers to what a normal prion protein does could help lead them to develop treatments and disease-prevention measures against human prion diseases, such as Creutzfeldt-Jakob disease, fatal familial insomnia and kuru, as well as animal prion diseases, such as scrapie in sheep and chronic wasting disease in cervids.
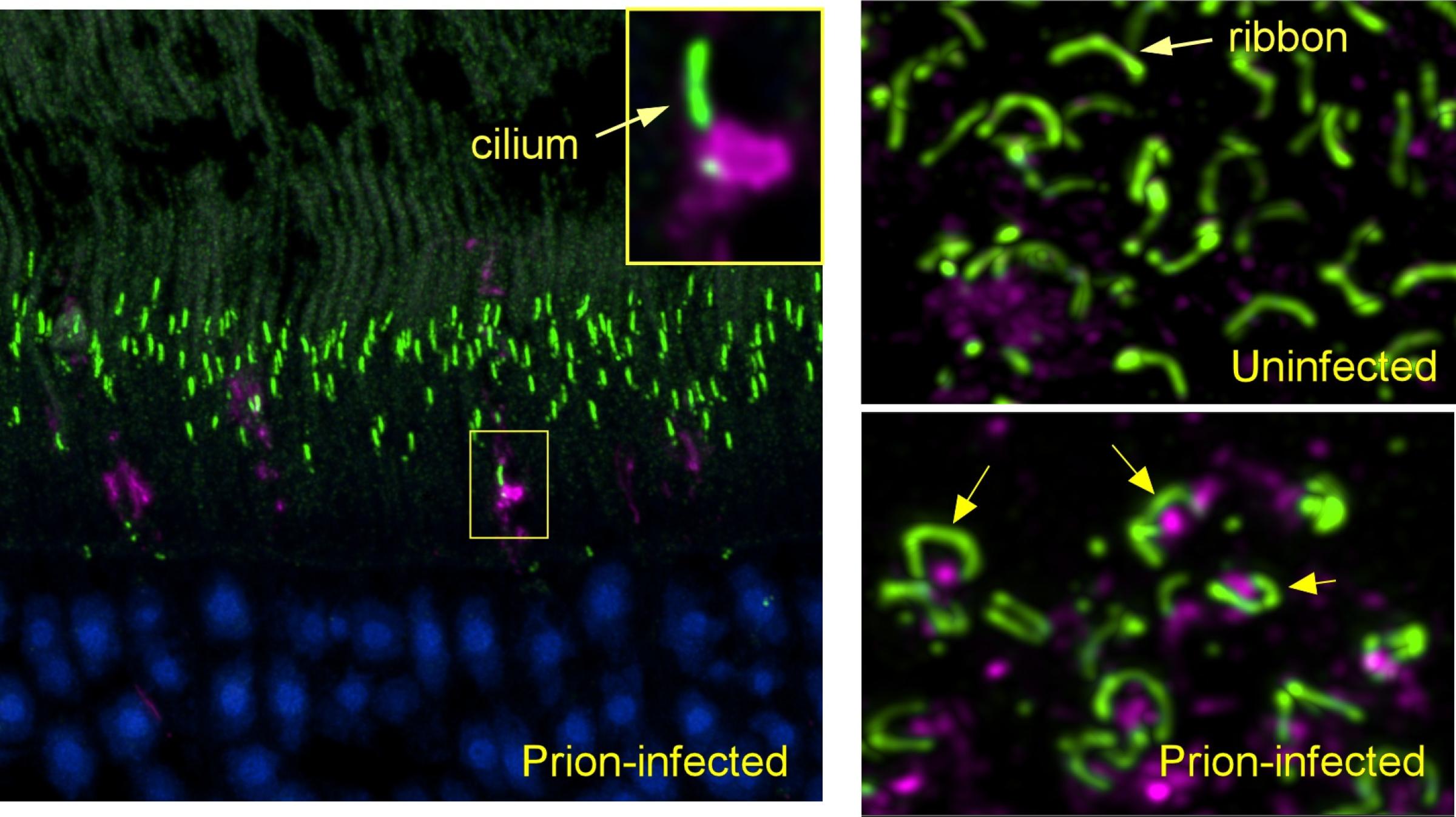
Scientists Identify Locations of Early Prion Protein Deposition in Retina
January 29, 2021
The earliest eye damage from prion disease takes place in the cone photoreceptor cells according to a new NIH study of prion protein accumulation.